FINCHES-online
Below, we provide some information detailing both technical/practical considerations and scientific interpretation of the results generated by FINCHES-online.
Technical information
What is FINCHES-online?
FINCHES-online is a web server that provides access to a subset of the functionality encoded in the Python package FINCHES. FINCES is a Python package developed to predict IDR-mediated intermolecular interactions using only sequence as an input.
FINCHES and FINCHES-online were developed by Garrett Ginell & Alex Holehouse in the Holehouse lab at Washington University in St. Louis. A preprint is forthcoming, and the citation will be:
Garrett M. Ginell, Ryan. J Emenecker, Jeffrey M. Lotthammer, Emery T. Usher, Alex S. Holehouse
Direct prediction of intermolecular interactions driven by disordered regions. bioRxiv (2024)
How can I cite FINCHES-online?
When citing FINCHES-online, please cite the preprint for the FINCHES package, along with the original force field paper you used. For example, if you used the Mpipi-GG implementation, a methods section/citation would look like this:
Intermolecular interactions were predicted using FINCHES-online with the Mpipi force field (Ginell et al., 2024; Joseph and Reinhardt et al., 2021).
We STRONGLY STRONGLY encourage you to cite the original force field papers when citing FINCHES. The accuracy of these underlying force fields is integral to FINCHES, and the developers of those force fields deserve credit. Similarly, if phase diagrams are predicted, we encourage you to cite both FINCHES and the original Qian et al. paper that derived the analytical solution we employ here. Full citations are provided at the end of this page.
How can I contact the developers?
For questions, comments, or bug reports, please raise an issue on the FINCHES GitHub page. Alternatively, feel free to reach out to Alex at Alex dot Holehouse (funny at sign) wustl dot edu.
Scientific information
What is FINCHES?
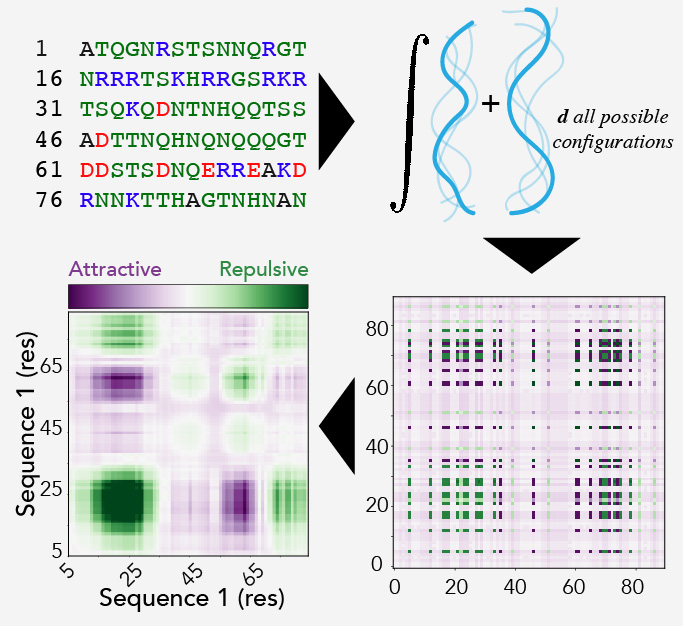 FINCHES implements a set of routines for converting parameters from coarse-grained force fields into mean-field predictions of intermolecular interactions. Broadly, this is done as follows:
FINCHES implements a set of routines for converting parameters from coarse-grained force fields into mean-field predictions of intermolecular interactions. Broadly, this is done as follows:
- Input: Two amino acid sequences
- Calculate the average interaction strength between all possible pairs of amino acids in the two sequences
- Modulate those average interaction strengths based on local sequence context (currently, this considers electrostatic and hydrophobic effects explicitly
- Using a smoothing function to average over local regions to identify subregions within the IDR that have the potential to facilitate attractive or repulsive interactions with a partner
- If selected, folded domain residues are then zeroed out, as they are not appropriately captured in this workflow in the absence of structural information
- Output: A predicted intermolecular interaction map (intermap) is generated and can be interactively interrogated or saved as a high-resolution PNG.
How should I interpret predictions made by FINCHES-online
The key thing - that we cannot emphasize more strongly - is that predictions made by FINCHES should be interpreted qualitatively or semi-quantitatively at best. Coarse-grained models make a lot of assumptions regarding intermolecular interactions. We then compound those assumptions by removing excluded volume effects and capturing cooperativity within an IDR at a very local level only. Consequently, the way to think about a FINCHES-based prediction is that subregions on an intermap that are deep purple should be thought of as regions that are likely to interact if they get the chance to . There are many reasons why those regions may NOT actually interact, not least due to competition with our regions (intra- or inter-molecularly) that will compete. Similarly, our ability to predict attractive and repulsive interactions is limited to chemical specificity. This means if an IDR interacts via a sequence-specific interface, we will NOT capture that. Put another way, we are sensitive to False Negatives if the mode of intermolecular interaction is driven by sequence-specific interactions. For intermap prediction, we envisage these predictions to facilitate a detailed, mechanistic investigation into the molecular interactions predicted here. FINCHES is particularly useful in the following situations:- When you know two proteins with large IDRs interact and would like to identify subregions that may contribute to this interaction and make predictions as to how mutations will alter interactions driven by those subregions.
- When you want to explain why two proteins do/do not interact
- When you're starting out on a project and would like to garner some intuition as to which IDR-containing protein(s) in a pathway or process may interact with one another
- When your protein undergoes phase separation, and you'd like to make targeted mutations to disrupt or enhance that behavior, combining intermap prediction with phase diagram prediction offers a route to make predictions regarding the underlying driving forces that enable an IDR to facilitate multivalent interactions needed for phase separation or condensate formation.
- For garnering intuition as to how changes in a sequence may be expected to shift a phase boundary (e.g., for interrogating the effects of mutations or post-translational modifications via PTM-mimicking mutations)
- For investigating if and how distinct isoforms may alter the phase separation propensity of a protein
Why are there two force field options?
FINCHES-online provides two force field options: Mpipi-GG and CALVADOS2.
Mpipi-GG is a finetuned version of the Mpipi (V1) force field, which itself was developed by Jerelle Joseph, Aleks Reinhardt, and Rosana Collepardo-Guevara (Joseph and Reinhardt et al., 2021). Mpipi-GG was reported by Lotthammer, Ginell, and Griffith et al. (2024). We encourage you to cite the original Mpipi paper if Mpipi-GG is used.
CALVADOS2 is an improved version of the CALVADOS force field, both of which were developed by Giulio Tesei and Kresten Lindorff-Larsen. CALVADOS2 was reported by (Tesei and Lindorff-Larsen, 2022). We encourage you to cite either the original CALVADOS paper or the more recent CALVADOS2 paper
Both force fields are reasonably good at capturing intra- and intermolecular IDR-mediated interactions. In our experience, CALVADOS2 does a better job capturing interactions driven by aliphatic residues. While quantitative agreement between predictions made with these two models is not always present, qualitative agreement—which is how FINCHES-based predictions should be used—is generally good.
Limitations and caveats
The limitations and caveats associated with FINCHES and FINCHES online are numerous and important. Please refer to the extensive discussion in the supplementary information of the forthcoming preprint for a detailed discussion of these topics, but we have done our best to calibrate expectations in this page.
Full citations
FINCHES paper
Garrett M. Ginell, Ryan. J Emenecker, Jeffrey M. Lotthammer, Emery T. Usher, Alex S. Holehouse
Direct prediction of intermolecular interactions driven by disordered regions. bioRxiv (2024)
Mpipi papers
Joseph, J. A., Reinhardt, A., Aguirre, A., Chew, P. Y., Russell, K. O., Espinosa, J. R., Garaizar, A. & Collepardo-Guevara, R.
Physics-driven coarse-grained model for biomolecular phase separation with near-quantitative accuracy.
Nat Comput Sci 1, 732–743 (2021).
Lotthammer, J. M., Ginell, G. M., Griffith, D., Emenecker, R. J. & Holehouse, A. S.
Direct prediction of intrinsically disordered protein conformational properties from sequence.
Nat. Methods 21, 465–476 (2024).
CALVADOS papers
Tesei, G., Schulze, T. K., Crehuet, R. & Lindorff-Larsen, K.
Accurate model of liquid-liquid phase behavior of intrinsically disordered proteins from optimization of single-chain properties.
Proc. Natl. Acad. Sci. U. S. A. 118, (2021).
Tesei, G. & Lindorff-Larsen, K.
Improved predictions of phase behaviour of intrinsically disordered proteins by tuning the interaction range.
Open Res. Eur. 2, 94 (2022).
Phase diagram prediction
Qian, D., Michaels, T. C. T. & Knowles, T. P. J.
Analytical Solution to the Flory-Huggins Model.
J. Phys. Chem. Lett. 13, 7853–7860 (2022).